Computed Structure Models
-
Introduction
Structural biology focuses on understanding the relationship between form (or shape) and functions. Since the early 1960s, researchers have developed and used experimental methods such as X-ray crystallography, Nuclear Magnetic Resonance, and Electron Microscopy to determine the three-dimensional (3D) atomic-level structures of proteins, nucleic acids, and their various complexes (Figure 2, left hand panel). The major motivation for these experiments is to explore and understand the molecular basis of biological function.
Experimentally-determined 3D structures of biological macromolecules are archived in the open-access Protein Data Bank (PDB) as managed by the Worldwide Protein Data Bank (wwPDB). These structural data are freely available from the PDB Archive and wwPDB member websites with no limitations on usage. Experimental structure determination is time consuming, expensive, and for many biomolecules it is technically challenging. Although there are billions of proteins in nature, experimental structures for only a few hundred thousand of them are currently available. This is where protein structure predictions by computational methods can help.
Protein Structure Prediction
Experiments in the early 1970s showed that proteins fold into functional shapes and that particular protein sequences spontaneously adopt characteristic folded 3D structures (Anfinsen, 1973). Today, much of our understanding of structural biology relies on a central principle: the sequence of amino acid residues in a protein chain determines its functional 3D shape. Experimentally-determined 3D structures of proteins have shed much light on protein sequence and structure relationships. Yet computational and experimental researchers have found it challenging to understand the precise steps involved in folding an unstructured protein in solution to its compact globular or other functional shape.
A native or stably-folded protein is thought to exist in the 3D conformation with lowest free energy. So protein structure prediction “computationally folds” polypeptide chains to form stabilizing intramolecular interactions that minimize the overall free energy of the molecule. In well-folded models, non-polar amino acid side chains typically face into the hydrophobic core of the protein, while polar and charged amino acid side chains typically face out from the hydrophobic core into the aqueous environment. Such calculations require prohibitive computing resources, so ab initio modeling or prediction of protein structure using just amino acid sequences and physics-based software tools (e.g., Rosetta) is currently only possible for small polypeptide chains. Computed structure models (or CSMs) of larger proteins need additional information for structure prediction. The following two approaches have been used:
Template-based modeling
Proteins with similar amino acid sequences (more than ~30% sequence identity) are known to fold into similar structures. So structure prediction of a new protein can be based on the known structure of a homologous protein (provided it has a sufficiently similar sequence). The success of this approach stems from the parsimonious nature of evolution, wherein successful 3D protein folds are reused. According to estimates, roughly 10,000 distinct polypeptide chain folds account for most naturally-occurring proteins. For many years, publicly-available computational services like Modeller/ModBase and ProMod3/SWISS-MODEL have used homology modeling and protein threading to predict protein structures. These approaches depend on finding an experimentally-determined protein structure with similar sequence to use as a modeling template or scaffold (Figure 1). Homology modeling is typically successful if a template with >40% sequence identity is available. Open access to 3D protein structure data has made this type of structure prediction broadly possible. The main idea is to use the many protein structures in the PDB as guides or templates for structure prediction.
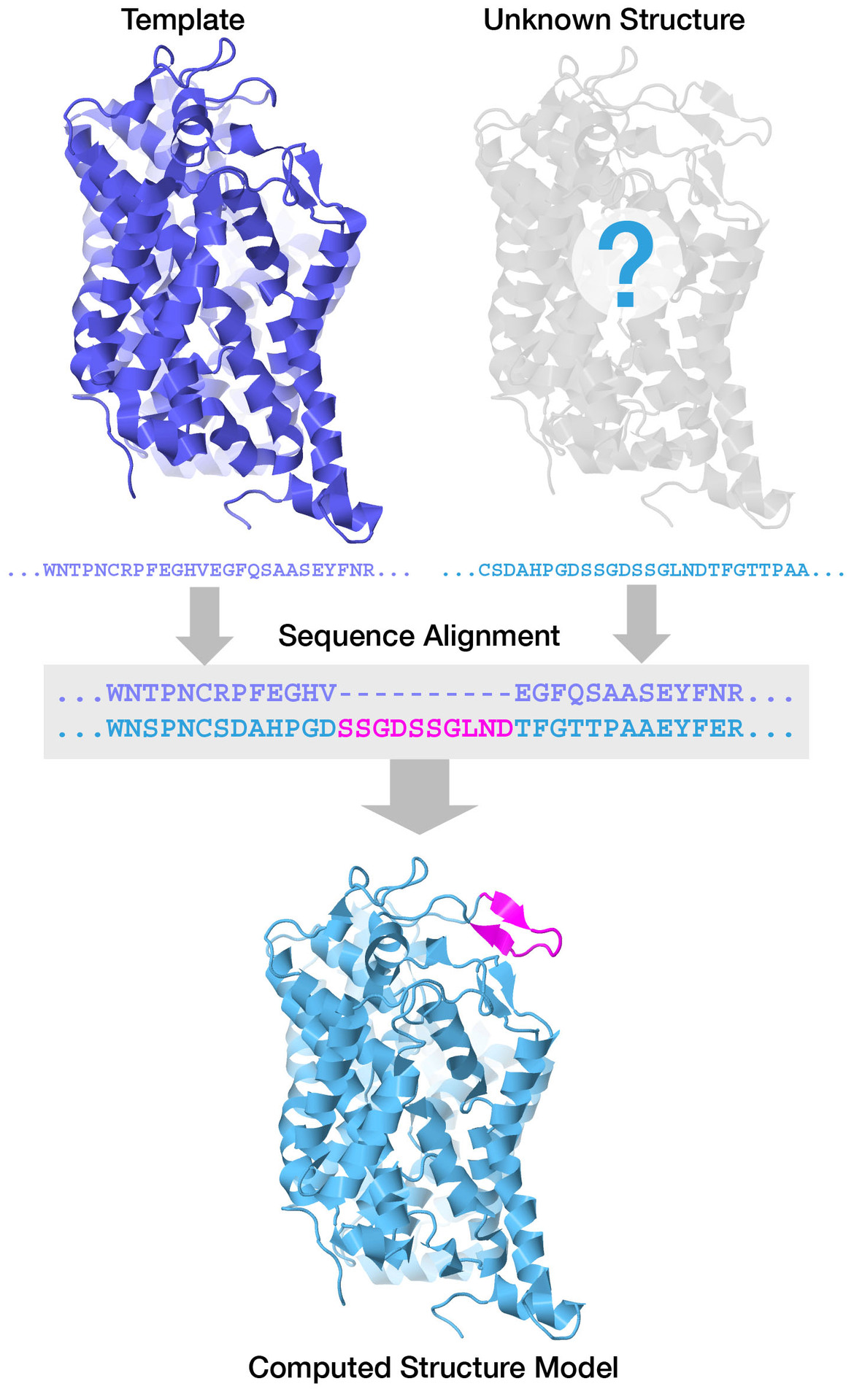
|
| Figure 1: Experimentally-determined structures may be used as modeling templates or scaffolds to predict structures of proteins with similar sequences. Here, the crystallographic structure of the dopamine transporter from fruit flies (upper left, PDB ID pdb_00004xp4) is used as a template to model the human protein. The sequence has 55% sequence identity with the human protein, but the sequence alignment reveals that one loop of the human protein is 10 amino acids longer than in fruit flies. The CSM of the human protein (bottom), generated by homology modeling at Swiss-Model, is very similar to the fruit fly protein, and includes modeled coordinates for this loop (magenta). |
Template-free modeling
When homologous structures (or templates) are not available, instead of relying on global homologies, local features of the structure are matched to build these models. Multiple sequence alignments can be used to identify evolutionarily conserved protein families. Access to vast amounts of sequencing and experimentally determined structural data show that in conserved protein families, amino acids that are in close proximity in 3D may be mutated so that their locations are exchanged but the interactions and physical distance between them is preserved. These correlated mutations or covariations can provide distance constraints and guide prediction of intramolecular contacts. Additionally, fragment matches and local secondary structure predictions can be included in template-free structure prediction (Figure 2, right hand panel). While advances in genome sequencing have given us access to vast amounts of protein sequence data needed for this approach, access to machine learning approaches has made such computations feasible.
Generating Computed Structure Models
Since the early 1990s, the Critical Assessment of Techniques for Protein Structure Prediction (CASP) team has organized biennial challenges to encourage development and improvement of computational algorithms for structure prediction. In 2020, AlphaFold2 (Jumper et al., 2021) showed that protein structures can be accurately predicted using artificial intelligence (AI) and machine learning (ML) approaches. Currently, AlphaFold2 and RoseTTAFold (Baek et al., 2021) are the most successful software tools for predicting protein structure from sequence. Both software tools build on decades of methodological research on structure prediction and rely on open access to the immense number of sequences in genomic sequence databases plus nearly two hundred thousand experimentally-determined 3D structures of proteins stored in the PDB.
AlphaFold2 generates computed structure models of proteins in a stepwise fashion. First, the sequence of the protein to be modeled is used to generate a multiple sequence alignment. Thereafter, an iterative process interrogates the multiple sequence alignment, refines the predicted 3D contacts within the polypeptide chain. Additional AI/ML processes then predict the 3D structure by breaking an initial protein model into individual amino acids and computationally recombining them in a way that is consistent with the predicted contacts. In the final model, a confidence score called “predicted local distance difference test” (pLDDT) is computed for each amino acid residue to estimate how well the method has converged - i.e., how well the predicted structure agrees with multiple sequence alignment data and PDB structure information (Jumper et al., 2021). pLDDT scores range from 0 to 100, with higher numbers indicating regions that have been confidently predicted versus those for which the prediction is less certain. The CSMs of compact globular proteins are comparable in accuracy to that of low-resolution experimental structures. Since these methods rely on experimental structures and sequence information, we expect that as the PDB continues to grow and computational tools get better, protein structure prediction accuracy will continue to improve.

|
| Figure 2: Experimental approaches and computation for determination of protein structures both rely on open access to genomic and 3D structure data. Here, methods for determining the structure of the RNA-binding protein Nova-2 are shown. The crystallographic structures (left) were determined for isolated domains of the structure, and reveal the atomic details of the interaction with RNA. The computed structure (right) includes the entire polypeptide chain, which is predicted to include three well-folded domains (blue) connected by unstructured linkers (in yellow and orange). Illustration by Maria Voigt. |
Using Computed Structure Models
Whenever a well-resolved 3D structure of a particular protein or protein domain is available from the PDB, it should be used preferentially instead of its corresponding CSM (Shao et al., 2022). Approximately 95% of the 3D structures in the PDB that have been determined using macromolecular crystallography are more accurate than corresponding CSMs generated using current AI/ML methods. When experimental structures are not available, CSMs can provide great starting points for structural explorations. At present, millions of CSMs generated using AI/ML methods are freely available for download from AlphaFoldDB (CSMs generated by Google DeepMind, Jumper et al., 2021, and Varadi et al., 2021), the ModelArchive (CSMs generated by RoseTTAFold, RoseTTAFold2, and AlphaFold2 users operating independently of DeepMind, Humphreys et al., 2021), and also from the RCSB PDB web portal RCSB.org. Learn more about CSMs and RCSB.org.
The pLDDT confidence score identifies the most trustworthy portions of the predicted structure. Regions with pLDDT greater than 0.7 (classified as very high or confident) often provide acceptable predictions of folded domains of proteins. Regions with lower pLDDT values can represent regions that aren’t structured in the protein, regions that play a role in higher-order protein assemblies, or untrustworthy hallucinations from the prediction method. For larger and/or multidomain proteins, CSMs should be analyzed carefully before they can be used to best effect. For example, see the example of Src protein presented below.
Computational protein structure prediction methods were optimized to predict 3D structures of proteins and protein domains that are well-folded, relatively inflexible, and do not form higher-order structures with other proteins. Notwithstanding these limitations, AlphaFold2 and RoseTTAfold/RoseTTAFold2 are so successful in predicting structures of well-ordered domains in proteins, they are showing utility in several applications. CSMs are being used:
- by molecular and cellular biologists to develop testable hypotheses regarding the functional importance of proteins, individual domains, or specific amino acids in a protein that are not present in the PDB (e.g., Panicum virgatum or switchgrass proteins for which there are only four structures present in the PDB).
- in structure-based drug discovery and design. Enzyme inhibitors and many drugs bind to conserved pockets or active sites that can be accurately predicted by AI/ML methods.
- to accelerate integrative structural biology. Predicted structures of individual proteins or domains may be fit into experimental maps of higher-order structures, building up our 3D structural knowledge of complex assemblies piece-by-piece.
Case Study: Computed Structure Model of the Src Oncoprotein
In the real world, protein structures are dynamic - they may adopt different conformations under different conditions, so their interactions and functions cannot be explained with just one 3D structure. The Src oncoprotein, named for its similarity to a Rous Sarcoma Virus protein, exemplifies both the power and the limitations of current AI/ML tools for protein structure prediction. Human Src is a multi-domain protein that consists of a flexible N-terminal tail, an SH3 domain, an SH2 domain, a protein kinase domain, and a small C-terminal tail. When the Src protein is active, the protein’s three domains are well separated from one another in space, allowing the SH2 and SH3 domains to interact with different binding partners, and precisely locate the protein kinase domain inside the cell to catalyze addition of phosphate groups to its target proteins.
The C-terminal tail of Src contains a key functional tyrosine amino acid residue (Tyr-529), which when phosphorylated by another protein kinase (known as C-terminal Src kinase or Csk, PDB pdb_00001byg) can downregulate Src protein’s activity. The SH2 domain binds phosphorylated Tyr-529 within the C-terminal tail and the SH3 domain binds to a proline-rich sequence connecting the SH2 domain and the protein kinase domain, thereby locking the entire polypeptide chain into a compact, closed conformation (Figure 3, left). An experimentally-determined structure (PDB pdb_00002src) revealed that the inactive form of Src protein is indeed compact.
Both AlphaFold2 and RoseTTAFold generate reasonably accurate CSMs for each of the three globular domains (accounting for most of the protein chain). However, they both incorrectly predict that the active, un-phosphorylated form of Src has three domains associated tightly with one another to adopt the conformation characteristic of the phosphorylated, inactive form of the enzyme (Figure 3, right). They also predict a long, unstructured N-terminal tail with low confidence, which is probably intrinsically-disordered.

|
| Figure 3: The crystallographic structure of the inactive conformation (left, PDB ID pdb_00002src) reveals the atomic details of the recognition of phosphotyrosine (red and pink spacefill) by the SH2 domain, but is missing the N-terminal tail. The crystallographic structure is colored to highlight the functional domains - Protein kinase (orange), SH2 domain in light blue, and SH3 domain in dark blue. Computed Structure Model of human Src oncogene protein predicted by AlphaFold2 (RCSB.org assigned ID AF_AFP12931F1, right), colored by confidence level (pLDDT scores). The model includes a compact folded structure with good confidence that corresponds to the inactive conformation of the protein, but with an unphosphorylated form of the tyrosine (pink). The model includes a long unstructured N-terminal tail with very low confidence. |
This real-world example underscores the importance of appreciating and understanding biological context and using experimentally-determined structures freely available from the PDB when working with CSMs. It also brings to mind the aphorism generally attributed to the famous statistician George Box, “All models are wrong, but some are useful."
Authors
David Goodsell, Shuchismita Dutta, and Stephen K. Burley
References
- Anfinsen, C. B. (1973). Principles that Govern the Folding of Protein Chains, Science, 181, 223-230, https://doi.org/10.1126/science.181.4096.223
- Baek, M., DiMaio, F., Anishchenko, I., Dauparas, J., Ovchinnikov, S., Lee, G. R., Wang, J., Cong, Q., Kinch, L. N., Schaeffer, R. D., Millán, C., Park, H., Adams, C., Glassman, C. R., DeGiovanni, A., Pereira, J. H., Rodrigues, A. V., van Dijk, A. A., Ebrecht, A. C., Opperman, D. J., … Baker, D. (2021). Accurate prediction of protein structures and interactions using a three-track neural network. Science, 373, 871–876. https://doi.org/10.1126/science.abj8754
- Berman, H., Henrick, K. & Nakamura, H. (2003). Announcing the worldwide Protein Data Bank. Nat Struct Mol Biol 10, 980. https://doi.org/10.1038/nsb1203-980
- Jumper, J., Evans, R., Pritzel, A. et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589. https://doi.org/10.1038/s41586-021-03819-2
- Humphreys, I. R., Pei, J., Baek, M., Krishnakumar, A., Anishchenko, I., Ovchinnikov, S., Zhang, J., Ness, T. J., Banjade, S., Bagde, S. R., Stancheva, V. G., Li, X. H., Liu, K., Zheng, Z., Barrero, D. J., Roy, U., Kuper, J., Fernández, I. S., Szakal, B., Branzei, D., … Baker, D. (2021). Computed structures of core eukaryotic protein complexes. Science, 374(6573), eabm4805. https://doi.org/10.1126/science.abm4805
- Shao, C., Bittrich, S., Wang, S., and Burley, S.K. (2022). “Assessing PDB Macromolecular Crystal Structure Confidence at the Individual Amino Acid Residue Level”, Structure. 30(10):1385-1394.e3. https://doi.org/10.1016/j.str.2022.08.004
- Varadi, M., Anyango, S., Deshpande, M., Nair, S., Natassia, C., Yordanova, G., Yuan, D., Stroe, O., Wood, G., Laydon, A., Žídek, A., Green, T., Tunyasuvunakool, K., Petersen, S., Jumper, J., Clancy, E., Green, R., Vora, A., Lutfi, M., Figurnov, M., Cowie, A., Hobbs, N., Kohli, P., Kleywegt, G., Birney, E., Hassabis, D., Velankar, S. (2022). AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models, Nucleic Acids Research, 50, D439–D444, https://doi.org/10.1093/nar/gkab1061
- wwPDB consortium. (2019). Protein Data Bank: the single global archive for 3D macromolecular structure data, Nucleic Acids Research, 47, D520–D528, https://doi.org/10.1093/nar/gky949
