Aminoglycoside Modifying Enzymes
Introduction
There are various mechanisms by which bacteria can become resistant to aminoglycosides - the most prevalent mechanism seen clinically is antibiotic inactivation by enzymatic modifications (Ramirez and Tolmasky, 2010). Aminoglycoside modifying enzymes catalyze the covalent modification of specific amino or hydroxyl groups in an antibiotic, altering its physicochemical properties. The chemically modified molecules have a low affinity for their target.
The uptake of aminoglycosides is energy-independent against a concentration gradient (Taber et al., 1987; Lang et al., 2023) - an initial ionic binding followed by two energy-dependent phases. Thus the modification of aminoglycosides also impacts drug uptake by the bacteria.
Source of Aminoglycoside Modifying Enzymes (AMEs)
Since there are several classes of AMEs the source of these enzymes are also diverse (Rather 1998). A few possible sources of AMEs are listed here:
* Some enzymes may have evolved alongside the aminoglycoside biosynthesis genes to protect the producers of these molecules from their toxic effects.
* Some enzymes may have been derived from housekeeping genes involved in cell metabolism - e.g., phosphorylation, adenylation, and acetylation.
* Some enzymes may be involved in metabolizing compounds e.g., peptidoglycans, lipopolysaccharides, and capsular polysaccharides.
The genes for these enzymes may have moved into plasmids and transposable elements and spread through plasmid exchange and dissemination of transposons. Under selective pressure of aminoglycosides, these mobile elements can overexpress the enzymes leading to drug resistance. Epidemiological studies demonstrate that diverse bacterial species with several distinct enzymes can modify almost every susceptible position in the aminoglycosides.
Types of AMEs
There are three families of aminoglycoside-modifying enzymes (AMEs, Figure 1) that decrease the affinity of aminoglycosides for their primary target, the 16S rRNA of the 30S ribosomal subunit. These three families of AMEs are:
* N-acetyltransferases (AACs) - these enzymes acetylate an amino group using acetyl-Coenzyme A
* O-nucleotidyltransferases (ANTs) - these enzymes transfer an adenyl group from ATP to a hydroxyl group of the antibiotic
* O-phosphotransferases (APHs) - these enzymes phosphorylate a hydroxyl group using either ATP or GTP
There are even bifunctional enzymes that combine the properties of two different families of AMEs, and they present a huge challenge for antimicrobial resistance currently.
The sites of modification of various classes of AMEs are shown in Figure 2.
![Figure 1: Schematic of aminoglycoside modifying enzyme classes. [AMP = adenosine monophosphate; NH2 = amino group; OH = hydroxyl group] (Ramirez & Tolmasky, 2010).](https://cdn.rcsb.org/rcsb-pdb/content/6382831166d6046810db7f19/ame-types.png)
|
| Figure 1: Schematic of aminoglycoside modifying enzyme classes. [AMP = adenosine monophosphate; NH2 = amino group; OH = hydroxyl group] (Ramirez & Tolmasky, 2010). |
|
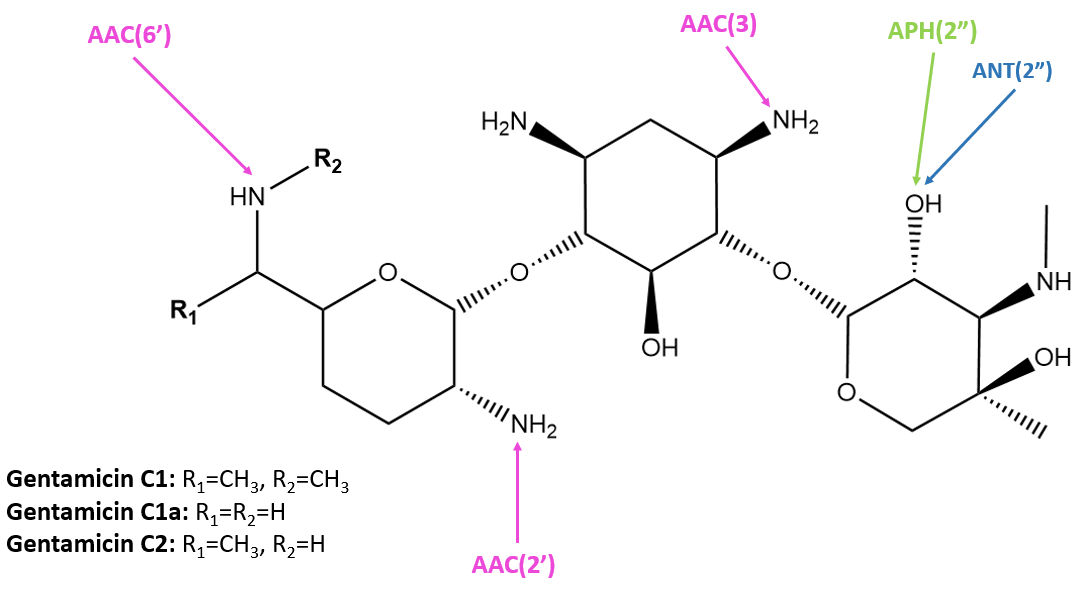
| Figure 2. Susceptible positions to AMEs in gentamicin (Mingeot-Leclercq and Tulkens, 1999). Structure was created with ChemDraw. |
N-acetyltransferases (AACs)
AACs make up the largest family of enzymes within the AMEs and are found both in gram-positive and gram-negative bacteria. They can add an acetyl group to the amine in a range of positions susceptible to modification to acetylate either nitrogen or oxygen group in the aminoglycoside. This consequently reduces the affinity of the antibiotic for the receptor 16S rRNA by 4 orders of magnitude (Mingeot-Leclercq and Tulkens, 1999).
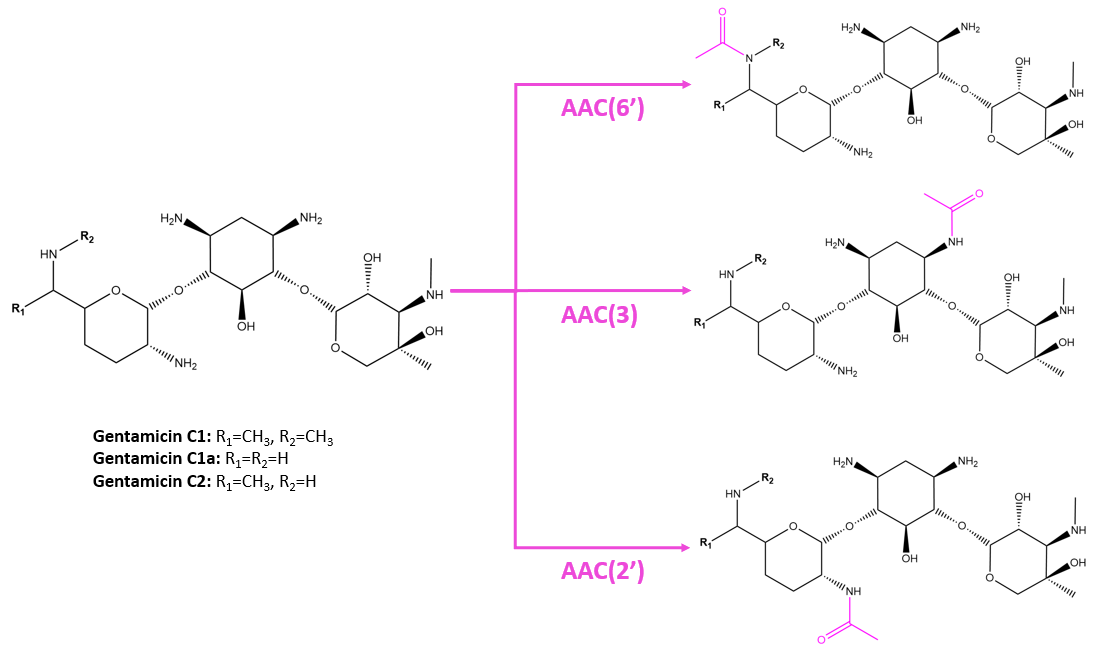
AAC(3) enzymes were the first to be described to confer resistance to gentamicin, kanamycin, and tobramycin. AAC(6′) enzymes are the most prevalent in clinical strains, conferring resistance to amikacin, gentamicin, kanamycin, neomycin, tobramycin, and more. AAC(2’) enzymes are encoded in Mycobacteria, and favor antibiotics with amine or hydroxyl groups at the 2’ position. AACs can have low amino acid sequence identity, but the overall structure is highly conserved. Modifications by AACs are shown in Figure 3.
|
| Figure 3. Modifications by AACs to gentamicin at different positions. Structures were created with ChemDraw. |
While AACs modify several antibiotics at the 6’, 2’, and 3’ positions, they are specific to the structures of different drugs. Mutagenesis studies show that differences in one amino acid of the AAC can change the specificity of the enzyme for certain drugs. For example, in Pseudomonas fluorescens, the aminoglycoside 6'-N-acetyltransferase type II enzyme can modify gentamicin to confer resistance but not to amikacin. Mutation of the Ser83 to Leu however allows the enzyme to confer resistance to amikacin but not to gentamicin (Lambert et al., 1994).
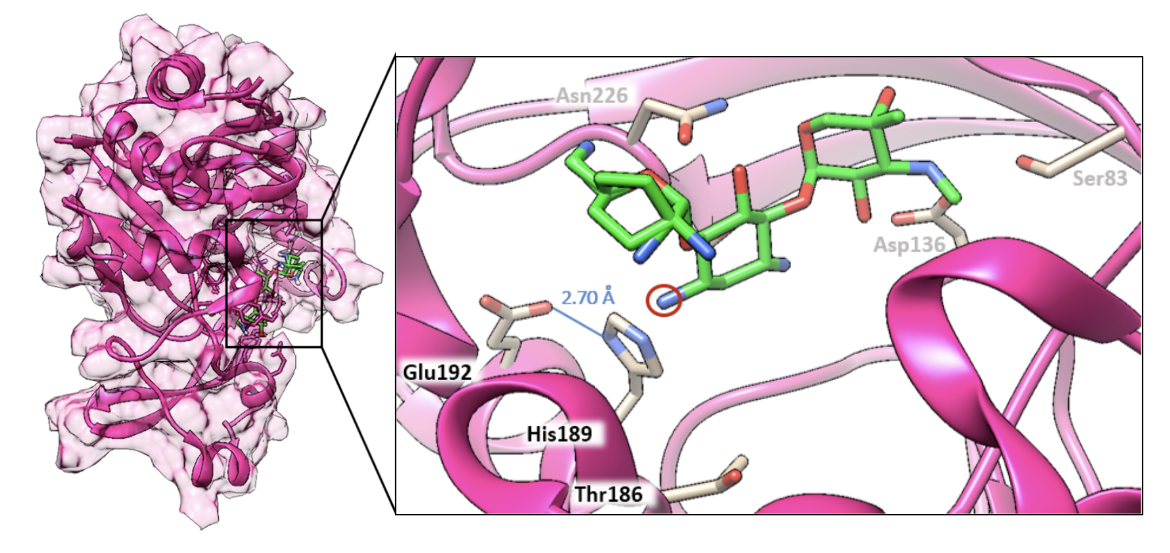
The structure of the enzyme aminoglycoside-N3-acetyltransferase, AAC(3)-VIa, bound to gentamicin (PDB ID 6np3; Kumar et al., 2019) is explored here in Figure 4.
|
| Figure 4. Structure of AAC(3)-VIa bound to gentamicin (PDB ID: 6np3; Kumar et al., 2019). Selected residues surrounding the antibiotic are labeled including the catalytic triad of Glu192, His189, and the N3 amine of gentamicin (circled in red). |
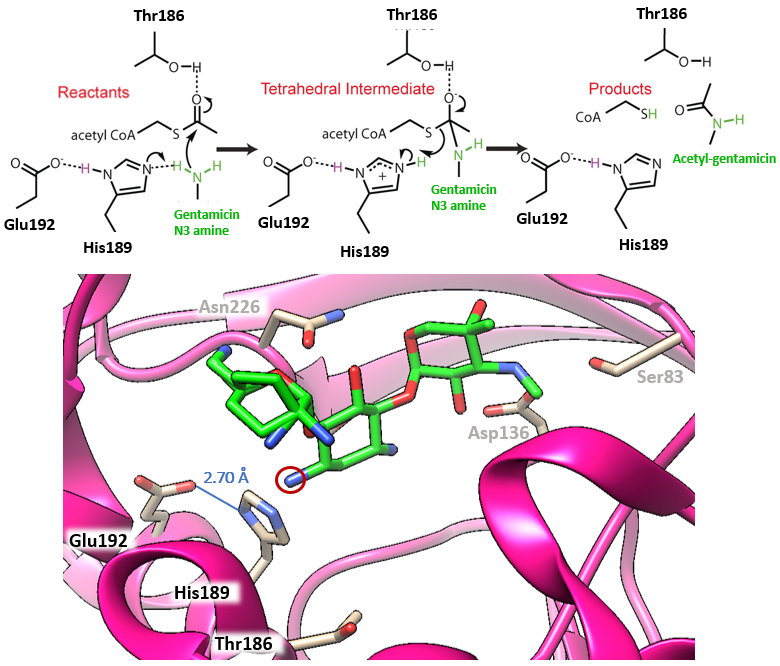
The mechanism of aminoglycoside-N3-acetyltransferase, AAC(3), in the context of AAC(3)-VIa is described herein. This enzyme uses acetyl coenzyme A as an acetyl donor as illustrated in Figure 5. The catalytic triad consists of Glu192, His189, and the N3 amine of the antibiotic. The hydrogen bond between Glu192 and His189 has a distance of 2.70 Å with the hydrogen being equidistant from each residue. This hydrogen bond is called a low-barrier hydrogen bond, a short hydrogen bond that is important for the catalytic efficiency of the enzyme. Due to the varying electrostatics of different antibiotic substrates, the pKa values of the catalytic triad can change and thus the hydrogen bond length can change, explaining why certain AACs have affinities from some antibiotics and not others. Gentamicin situates itself in the active site such that the hydrogen bond is in an ideal position for the mechanism of acetylation to occur.
|
| Figure 5. Mechanism of acetylation with gentamicin bound to the active site of AAC-VIa. The mechanism is color-coded to match the figure of the structure below. Mechanism adapted from Kumar et al., 2019). The catalytic triad consists of Glu192, His189, and the N3 amine of gentamicin (circled in red). The low-barrier hydrogen bond is labeled (PDB ID: 6np3; Kumar et al., 2019). |
O-phosphotransferases (APHs)
APHs make up the second largest family of enzymes within the AMEs (Krause et al., 2016). These enzymes are structurally and functionally similar to eukaryotic serine, threonine, and tyrosine kinases. They catalyze nucleotide-dependent phosphorylation of hydroxyl groups at various positions in aminoglycosides. A majority of the APH enzymes belong to the APH(3′) and (2") subfamilies. Interestingly the APH(2") aminoglycoside kinase family are unique in their ability to utilize GTP (instead of ATP) as a cofactor for antibiotic modification (Smith et al., 2012). Some APHs can use both ATP and GTP under physiological conditions (Shi and Berghuis, 2012)
Here we examine the structures of two APH enzymes:
* APH(2") aminoglycoside kinase, in complex with GDP and kanamycin (PDB ID 6ctz, Smith et al., 2019 - Figure 6)
* APH(9)-Ia, aminoglycoside kinase, in complex with ADP and Spectinomcyin (PDB ID 3i0o, Fong et al., 2010 - Figure 7)
The structure of APH(2")-IIIa is C-shaped and organized into three domains (Figure 6A) - an N-terminal domain (top), the central core domain (left) and the helical subdomain (bottom) (Smith et al., 2019). The nucleotide (that donates the phosphate binds between the N-terminal and core domains, and interacts with specific backbone atoms of a loop connecting the two domains (Figure 6B). The antibiotic sits between the core and helical domains, and interacts with a series of negatively charged amino acid side chains (Figure 6C). This 1.34 A˚ resolution structure represents a catalytically inactive abortive complex since the phosphate group that would be transferred to the antibiotic is missing.
A closeup of the GDP binding site (Figure 6B) shows that the Guanine base interacts with a few backbone atoms of residues in a loop connecting the N-terminal and central domains. The antibiotic binding site is located between the core and helical domains and lined by a series of negatively charged amino acids (Figure 6C). Kanamycin positions itself in the binding pocket to receive the phosphate group at the hydroxyl marked with the black arrowhead.
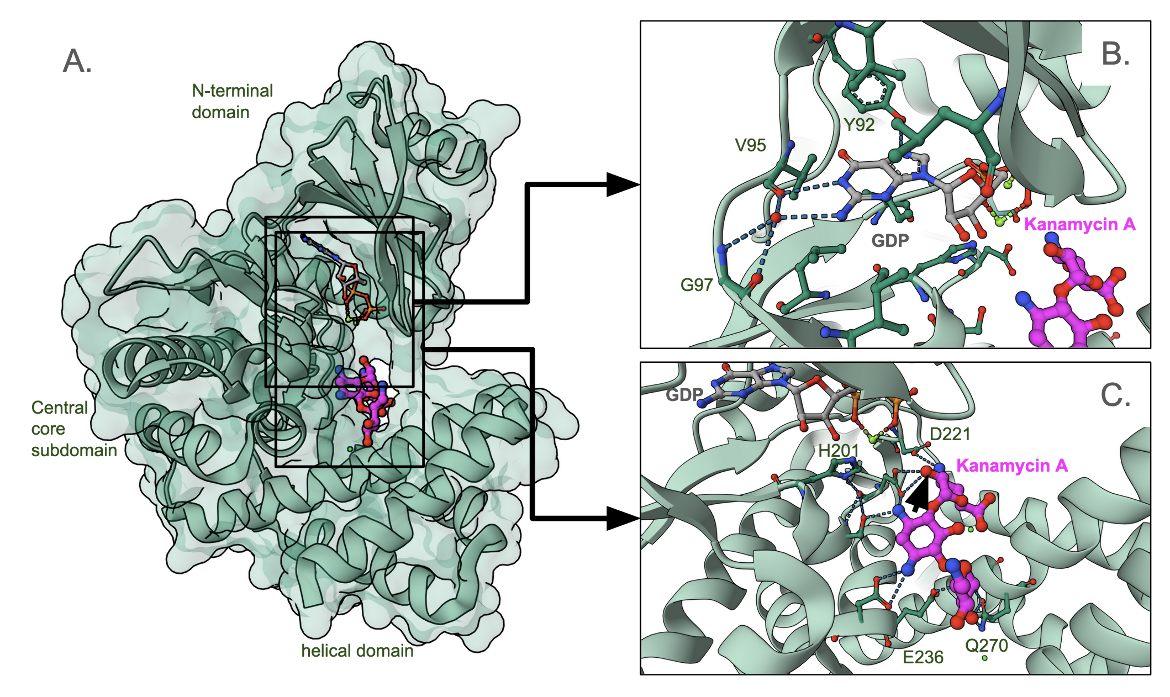
|
| Figure 6. APH(2′′) aminoglycoside kinase, in complex with GDP and kanamycin (PDB ID 6ctz, Smith et al., 2019 - Figure 6) A. Complete structure; B. a closeup of the GDP binding site. The carbon atoms of GDP are colored grey; C. a closeup of the antibiotic binding site. Carbon atoms of kanamycin are shown in magenta. |
The APH(9)-Ia is a C-shaped molecule with a β-strand-rich N-terminal lobe and an α-helix-rich C-terminal lobe (Figure 7A). The nucleotide that donates its terminal phosphate group (ATP in this case) is positioned closer to the N-terminal lobe (Figure 7B). The structure shown here includes an ADP so there is no terminal phosphate group seen here. The base (adenine) makes a few hydrogen binds with the backbone atoms of the linked between the N- and C-terminal lobes. The base also forms some pi-stacking interactions with Phe30. The antibiotic binds closer to the C-terminal lobe in a pocket lined by several polar and charged amino acids (Figure 7C). It is positioned to receive a phosphate group from ATP at the position marked by the black arrowhead.
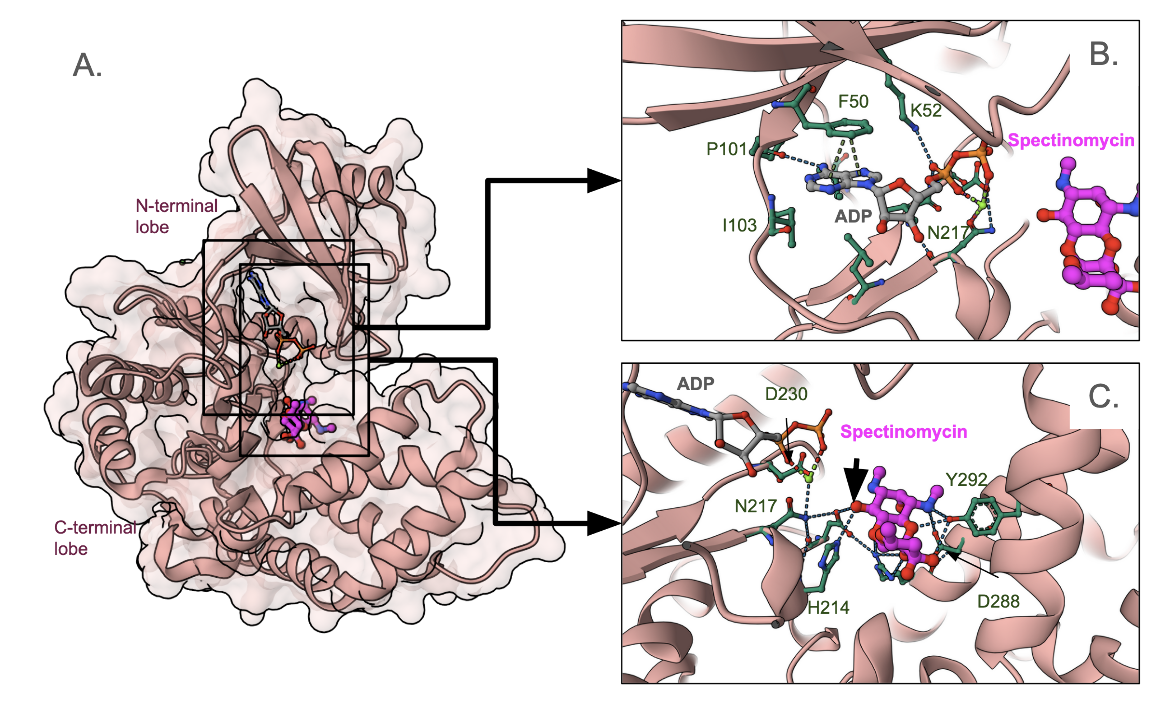
|
| Figure 7. APH(9)-Ia, aminoglycoside kinase, in complex with ADP and Spectinomcyin (PDB ID 3i0o, Fong et al., 2010). A. Complete structure; B. a closeup of the ADP binding site. The carbon atoms of ADP are colored grey; C. a closeup of the antibiotic binding site. Carbon atoms of spectinomycin are shown in magenta. |
O-nucleotidyltransferases (ANTs)
ANTs inactivate aminoglycosides by catalyzing the transfer of an AMP group from ATP to a hydroxyl group in the aminoglycoside molecule. There are five classes of ANTs, catalyzing the addition of AMP from an ATP donor to hydroxyl groups at positions 6, 9, 4', 2", and 3" (Ramires and Tolmasky, 2010). Here we examine the structure of an aminoglycoside 2″-nucleotidyltransferase [ANT(2″)] bound to tobramycin and an ATP analog (PDB ID 5cfs, Bassenden et al., 2016) in Figure 8.
Overall the structure of the ANT(2") enzyme is C-shaped. The aminoglycoside substrate and the cofactor (ATP) both bind in the active site such that they are surrounded by a large network of hydrogen bonds. Here AMPCPP (α−β non-hydrolyzable analogue of ATP) is used to form a complex. Here the phosphate groups are coordinated by two Mn2+ ions and a number of acid amino acids surround the active site - Asp44, Asp86, Glu88, and Asp131. The modification (adenylation) occurs at the O2" position of the antibiotic (marked with a black arrowhead in Figure 8B).
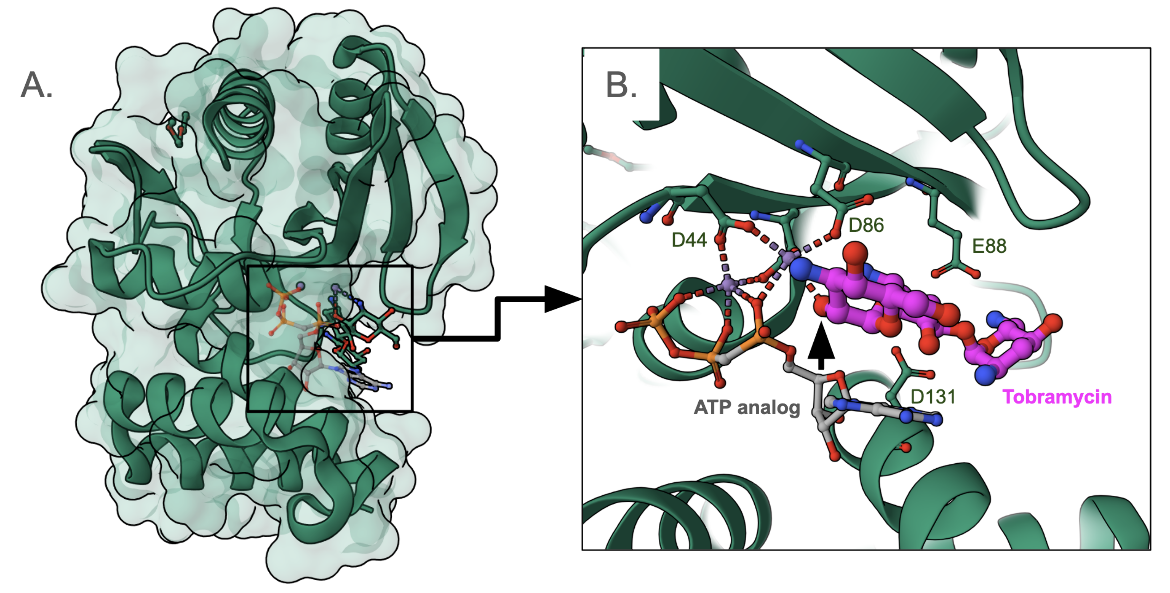
|
| Figure 8. Structure of ANT(2")-Ia in complex with an ATP analog (AMPCPP) and tobramycin, PDB ID 5cfs, Bassenden et al., 2016. The inset shows a closeup of the antibiotic binding and key residues in the binding site. The carbon atoms of tobramycin are colored magenta, while that of the ATP analog are colored grey. |
Clinical Implications of Resistance by AMEs
Aminoglycoside-modifying enzyme-encoding genes are highly prevalent among Pseudomonas aeruginosa clinical isolates worldwide (Ahmadian et al., 2021). Many clinical strains of K. pneumoniae and A. baumannii isolates from around the world also have been found to have many AMEs. However, the presence of enzymes is not sufficient to explain antibiotic resistance rates (Zhang et al., 2023). Therefore, other factors leading to resistance should be investigated and considered in developing treatment plans.
References
Ahmadian, L., Norouzi Bazgir, Z., Ahanjan, M., Valadan, R., Goli, H. R. (2021) Role of Aminoglycoside-Modifying Enzymes (AMEs) in Resistance to Aminoglycosides among Clinical Isolates of Pseudomonas aeruginosa in the North of Iran. Biomed Res Int. 2021:7077344. https://doi.org/10.1155/2021/7077344
Bassenden, A. V., Rodionov, D., Shi, K., Berghuis, A. M. (2016) Structural Analysis of the Tobramycin and Gentamicin Clinical Resistome Reveals Limitations for Next-generation Aminoglycoside Design. ACS Chem Biol. 11(5):1339-46. https://doi.org/10.1021/acschembio.5b01070
Fong, D. H., Lemke, C. T., Hwang, J., Xiong, B., Berghuis, A. M. (2010) Structure of the antibiotic resistance factor spectinomycin phosphotransferase from Legionella pneumophila. J Biol Chem. 285(13):9545-9555. https://doi.org/10.1074/jbc.m109.038364
Krause, K. M., Serio, A. W., Kane, T. R., Connolly, L. E. (2016) Aminoglycosides: An Overview. Cold Spring Harb Perspect Med. 6(6):a027029. https://doi.org/10.1101/cshperspect.a027029
Kumar, P., Agarwal, P. K., Waddell, M. B., Mittag, T., Serpersu, E. H., Cuneo, M. J. (2019) Low-Barrier and Canonical Hydrogen Bonds Modulate Activity and Specificity of a Catalytic Triad. Angew Chem Int Ed Engl. 58(45):16260-16266. https://doi.org/10.1002/anie.201908535
Lang, M., Carvalho, A., Baharoglu, Z., Mazel, D. (2023). Aminoglycoside uptake, stress, and potentiation in Gram-negative bacteria: new therapies with old molecules. Microbiol Mol Biol Rev 87:e00036-22. https://doi.org/10.1128/mmbr.00036-22
Lambert, T., Ploy, M. C., Courvalin, P. (1994) A spontaneous point mutation in the aac(6')-Ib' gene results in altered substrate specificity of aminoglycoside 6'-N-acetyltransferase of a Pseudomonas fluorescens strain. FEMS Microbiol Lett. 115(2-3):297-304. https://doi.org/10.1111/j.1574-6968.1994.tb06654.x
Mingeot-Leclercq, M. P., Tulkens, P. M. (1999) Aminoglycosides: nephrotoxicity. Antimicrob Agents Chemother. 43(5):1003-12. https://doi.org/10.1128/aac.43.5.1003
Ramirez, M. S., Tolmasky, M. E. (2010) Aminoglycoside modifying enzymes. Drug Resist Updat. 2010 Dec;13(6):151-71. https://doi.org/10.1016/j.drup.2010.08.003
Rather, P. N. (1981) Origins of the aminoglycoside modifying enzymes. Drug Resist Updat. 1(5):285-91. https://doi.org/10.1016/s1368-7646(98)80044-7
Shi, K., Berghuis, A. M. (2012) Structural basis for dual nucleotide selectivity of aminoglycoside 2''-phosphotransferase IVa provides insight on determinants of nucleotide specificity of aminoglycoside kinases. J Biol Chem. 287(16):13094-102. https://doi.org/10.1074/jbc.m112.349670
Smith, C. A., Toth, M., Frase, H., Byrnes, L. J., Vakulenko, S. B. (2012) Aminoglycoside 2''-phosphotransferase IIIa (APH(2'')-IIIa) prefers GTP over ATP: structural templates for nucleotide recognition in the bacterial aminoglycoside-2'' kinases. J Biol Chem. 287(16):12893-903. https://doi.org/10.1074/jbc.M112.341206
Smith, C. A., Toth, M., Stewart, N. K., Maltz, L., Vakulenko, S. B. (2019) Structural basis for the diversity of the mechanism of nucleotide hydrolysis by the aminoglycoside-2''-phosphotransferases. Acta Crystallogr D Struct Biol. 75(Pt 12):1129-1137. https://doi.org/10.1107/s2059798319015079
Taber, H. W., Mueller, J. P., Miller, P. F., Arrow, A. S. (1987) Bacterial uptake of aminoglycoside antibiotics. Microbiol Rev. 51(4):439-57. https://doi.org/10.1128/mr.51.4.439-457.1987
Zhang, Y., Zhang, N., Wang, M., Luo, M., Peng, Y., Li, Z., Xu, J., Ou, M., Kan, B., Li, X., Lu, X. (2023) The prevalence and distribution of aminoglycoside resistance genes,
Biosafety and Health, 5 (1),14-20. https://doi.org/10.1016/j.bsheal.2023.01.001
April 2025, Helen Gao, Shuchismita Dutta; Reviewed by Drs. Albert Berghuis and Tolou Golkar
https://doi.org/10.2210/rcsb_pdb/GH/AMR/drugs/amr-mech/AME
