Beta lactamase
Introduction
Since its discovery by Alexander Fleming in the late 1920s, penicillin (a molecule that contains a β-lactam ring) has become the most widely used class of antibiotics in modern medicine. Resistance to β-lactam antibiotics threatens its role in the successful treatment of infection. Therefore new generations of β-lactams are developed to increase the spectrum of bacteria that can be killed or to address drug resistance to older generations of β-lactam antibiotics (Bush and Bradford, 2016). In this past century, thousands of derivatives and new classes of β-lactam antibiotics have been discovered. Amoxicillin, a β-lactam antibiotic that has become a household name, is commonly used to treat infections, such as strep throat and urinary tract infections. Cephalexin is an antibiotic that is part of a newer class of β-lactams called cephalosporins and is commonly used to treat ear and respiratory tract infections. Learn more about β-lactams.
The most common way bacteria confer resistance to β-lactams is through the enzymes called β-lactamases. The idea that β-lactamases may have arisen from D-alanyl-D-alanine-cleaving peptidase, essential for the last stages of peptidoglycan synthesis was proposed in the 1960s (Tipper and Strominger, 1965). This was verified by comparing the structures of D-alanyl-D-alanine-peptidase and penicillin-hydrolyzing β-lactamase enzyme. Studies showed that the spatial arrangement of secondary structural elements and the site of penicillin binding in one enzyme was equivalent to that of the other (Kelly et al., 1986). Thus, the cell wall transpeptidases were the likely ancestor of the β-lactamases.
Thousands of different β-lactamase enzymes have emerged to combat the effects of β-lactam antibiotics. This article will describe the structures and functions of different classes of β-lactamase enzymes and their interactions with β-lactam drugs.
Normal Functions of β-lactams
β-lactams inactivate the penicillin-binding proteins of bacteria, thereby inhibiting the last stage of the peptidoglycan cell wall synthesis (i.e., the inhibition of transpeptidation) (Tipper, 1985). All β-lactam antibiotics have a β-lactam ring, a highly strained and reactive four-member ring containing an amide group covalently linked to a carbonyl carbon atom and another carbon atom in a β position (i.e., two carbon atoms away) with reference to the carbonyl carbon.
While the nitrogen atom of the amide group is single-bonded to a carbonyl carbon atom, the carbonyl group consists of a carbon double-bonded to an oxygen atom (Figure 1).
There are five subtypes of β-lactams which differ by the structure around the core β-lactam ring; these categories are penam, penem, carbapenem, cefem, and monobactams (Fernandes, Amador, and Prudêncio, 2013). Learn more about β-lactam antibiotics and their targets.
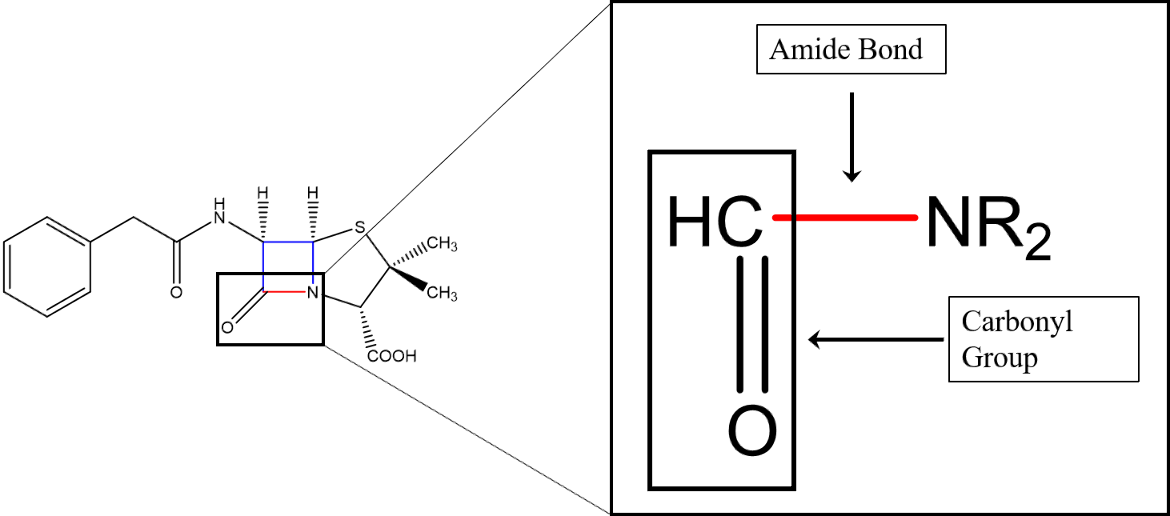
|
| Figure 1. The structural formula of Penicillin G with the four-membered β-lactam ring colored in red and blue. The amide bond that is enzymatically broken is colored in red. The critical amide functional group is magnified: the amine nitrogen is bonded to a carbonyl carbon; the R groups on an amine can be other carbon structures or hydrogen atoms. The structure was created using ChemDraw. |
Resistance Through β-lactamases
β-lactamases are enzymes that have evolved in bacteria that can modify antimicrobial molecules. Organisms, such as fungi, have been coexisting with bacteria for millennia. They have evolved mechanisms such as β-lactam antibiotic molecules to kill the bacteria they compete with. In response, bacteria, with their high degree of genetic plasticity, have evolved to produce β-lactamase enzymes to destroy β-lactam antibiotic molecules. Specifically, they break the amide bond within the β-lactam antibiotic molecule (shown in Figure 1).
β-lactamase enzymes are found in both gram-negative and gram-positive bacteria. β-lactamases inactivate penicillins and cephalosporins (cephems) by breaking the amide bond (shown in red in Figure 1) and opening the β-lactam ring. Genes encoding β-lactamases are named bla followed by the specific enzyme name. Located on transmittable genetic elements, these genes contribute to the high prevalence of β-lactamase-mediated resistance in the clinical setting (Munita and Arias, 2016).
After penicillin became widely available for clinical use, penicillin-resistant S. aureus rapidly emerged with plasmid-encoded penicillinase (penicillin-destroying) enzymes. These plasmids were readily transmitted between S. aureus strains which resulted in rapid dissemination of the resistance genes. New β-lactam compounds, namely ampicillin, were developed and had less susceptibility to these penicillinases (Munita and Arias, 2016). However, in 1965, a new plasmid-encoded β-lactamase capable of hydrolyzing ampicillin was discovered in Escherichia coli isolates from a patient named Temoneira in Athens, Greece. This β-lactamase was named after the patient with the designation TEM-1 (Paterson and Bonomo, 2005). In this pattern, the generation of new β-lactam drugs was regularly followed by the appearance of enzymes capable of destroying them. As a result, over 1,000 different β-lactamases have been described in scientific literature. The Ambler classification system has become the most widely used system to identify and describe the numerous β-lactamase enzymes. The Ambler system categorizes enzymes into A, B, C, and D categories (Munita and Arias, 2016). Explore the β-lactamases in the Comprehensive Antibiotic Resistance Database.
When initially proposed, the Ambler classifications separated β-lactamases into A and B groups, based on their molecular phylogeny due to amino acid sequence differences. It was then proposed that Class A β-lactamase enzymes diverged from a single ancestral gene (see Figure 2) and used a catalytic serine residue to break the amide bond of the β-lactam ring. Class B β-lactamases had no sequence similarity with the enzymes in class A (known at the time). They catalyzed β-lactam destruction using metal cofactors and a completely separate enzymatic pathway, so they were grouped into a new group called class B (Ambler, 1980). Later on, classes C and D were created, both of which include serine β-lactamases, but with little sequence similarity to each other. However, the structural similarities between these classes of enzymes indicate that they have descended from a common ancestor (Hall and Barlow, 2005). So, Ambler classes A, C, and D are all serine β-Lactamases (SBLs), that utilize a catalytic serine residue to destroy the β-Lactam drug (Munita and Arias, 2016).
Amino acid motifs are amino acid patterns that generate specific functions found in enzymes. Critical in SBLs is the motif SXXK, or serine - X - X - lysine, where X can be any residue. This motif contains lysine and serine; lysine commonly acts as a base that deprotonates the serine amino acid. This deprotonation enhances serine's reactivity and improves its ability to open the β-lactam ring (King et al., 2015).
Penicilin-binding proteins (PBPs) are necessary for bacterial survival. β-Lactam containing antibiotics bind to PBPs to deactivate them. The β-Lactamase enzymes bind to and deactivate the β-Lactam ring of antibiotics rendering them ineffective in binding to PBPs. In SBLs, the same acyl intermediate is formed showing that SBLs undergo very similar catalytic mechanisms. This intermediate is not found in class B β-Lactamases showing that class B enzymes utilize a separate catalytic mechanism. The SBLs are differentiated by the different orientations of the active site residues (Fernandes, Amador, and Prudêncio, 2013) as well as their amino acid sequences (Munita and Arias, 2016).
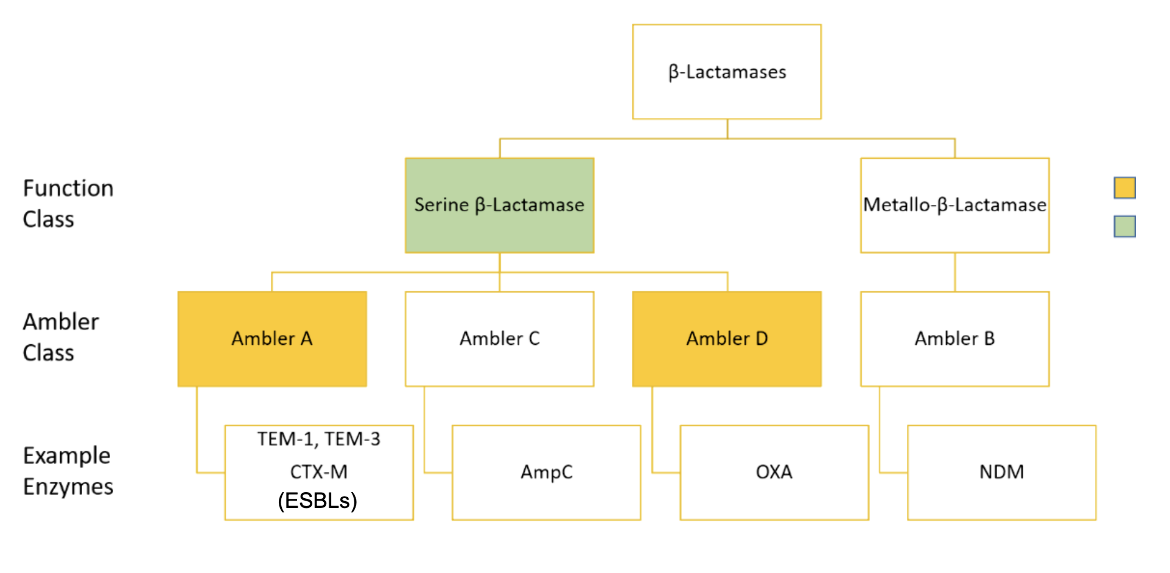
|
| Figure 2. Overview of the Ambler classification system (ESBL = extended spectrum β-lactamases). |
Ambler A
Ambler class A includes a wide range of serine β-Lactamases. Examples of clinically relevant β-Lactamase enzymes are TEM-1, CTX-M, and KPC ( or Klebsiella pneumoniae carbapenemase). The diversity of the class A β-Lactamases is a result of the antibiotics used against SBL-expressing bacteria. For example, resistance conferred by the TEM-1 β-Lactamase enzyme was combated by using extended-spectrum cephalosporin drugs, which are β-Lactam antibiotics but are effective against an extended range of bacteria. Following the use of these cephalosporins, extended-spectrum β-Lactamase (ESBL) mutants quickly evolved. These enzymes could destroy a wider range of β-Lactam antibiotics, hence their designation as "extended-spectrum." In this fashion, many class A SBLs evolved (Fernandes, Amador, and Prudêncio, 2013; Munita and Arias, 2016). Of great clinical importance are the CTX-M ESBLs. These β-Lactamases were named for their enhanced activity against the third-generation cephalosporin cefotaxime (Jia et al., 2017). CTX-M genes have been found on many kinds of mobile genetic elements that can be transmitted to other bacteria. Consequently, CTX-M genes have become the most prevalent ESBL and have greater potential to spread beyond hospital environments compared to other β-Lactamases (Munita and Arias, 2016; Fernandes, Amador, and Prudêncio, 2013).
The β-Lactamase mechanism can best be understood with TEM-1 as an example. The TEM class of enzymes is named after Temoniera, the first patient from whom the resistance enzyme was isolated in 1963 (Ruiz J. 2018). The TEM-1 enzyme is highly prevalent in bacteria and can destroy penicillins and first-generation cephalosporins (see Table 1).
Table 1. TEM-1 Data: Drug classes that TEM-1 β-Lactamase successfully resists and the bacterial species that possess genes expressing TEM-1. Information obtained from CARD.
| Drug Classes | Bacterial Species |
|---|---|
| Penem, Cephalosporin, Monobactam, Penam | Acinetobacter baumannii, Acinetobacter nosocomialis, Chlamydia trachomatis, Citrobacter amalonaticus, Citrobacter freundii, Citrobacter koseri, Enterobacter aerogenes, Enterobacter asburiae, Enterobacter cloacae, Enterobacter hormaechei, Enterobacter kobei, Escherichia coli, Haemophilus influenzae, Haemophilus parainfluenzae, Klebsiella oxytoca, Klebsiella pneumoniae, Morganella morganii, Neisseria gonorrhoeae, Proteus mirabilis, Providencia rettgeri, Providencia stuartii, Pseudomonas aeruginosa, Pseudomonas putida, Salmonella enterica, Serratia marcescens, Shigella dysenteriae, Shigella flexneri, Shigella sonnei, Vibrio cholerae |
The structure of TEM-1 consists of 2 domains (see Figure 3). Domain 1 contains a 5-stranded β-sheet covered by 3 ⍺-helices on one face and 1 ⍺-helix on the other. Domain 2 consists mostly of ⍺ helices, and the active site is located at the interface of these 2 domains (Fonzé et al., 1995). TEM-1 in E. coli contains the critical motif of Ser70, Thr71, Phe72, Lys73 (SXXK) which contains the serine and lysine needed for catalytic activity. As discussed earlier, Lys73 deprotonates the Oˠ (Gamma-Oxygen) of Ser70, enhancing its reactivity. Cleavage of the amide bond occurs through 2 fundamental steps: acylation and deacylation.
![Figure 3. TEM-1 β-Lactamase enzyme of E. coli. The Oˠ (gamma-oxygen) is located at the end of the serine residue and is colored red. Ser70 and Lys73 are part of the essential SXXK motif. [PDB ID: 1FQG] (Nitrogen - blue; Sulfur - yellow; Oxygen - red; Carbon - grey)](https://cdn.rcsb.org/rcsb-pdb/content/61b762c769569d045d35a99c/tem-1figure.png)
|
| Figure 3. TEM-1 β-Lactamase enzyme of E. coli. The Oˠ (gamma-oxygen) is located at the end of the serine residue and is colored red. Ser70 and Lys73 are part of the essential SXXK motif. [PDB ID: 1FQG] (Nitrogen - blue; Sulfur - yellow; Oxygen - red; Carbon - grey) |
Acylation of the β-Lactam ring to form an acyl intermediate is initiated by the attack of the Oˠ of the serine-70 residue in the TEM-1 β-Lactamase enzyme (Figure 4, Figure 5). This oxygen atom attacks the carbonyl carbon on the β-Lactam ring. This initiates a cascade of proton transfers, ultimately resulting in the cleavage of the amide bond (Strynadka et al., 1992).

|
| Figure 4. Condensed mechanism of acylation. Tetrahedral intermediates and proton-carrying residues have been excluded for simplicity. Arrows indicate electron pushing. The structures were created using ChemDraw (Strynadka et al., 1992). |
![Figure 5. The complex of penicillin G with the acyl intermediate of the TEM-1 β-Lactamase. The broken bond between the carbonyl carbon and the nitrogen of the β-Lactam ring is shown with the grey dashed line. [PDB: 1FQG] (Nitrogen - blue; Sulfur - yellow; Oxygen - red; Carbon - grey)](https://cdn.rcsb.org/rcsb-pdb/content/61b762c769569d045d35a99c/penn-acyl.png)
|
| Figure 5. The complex of penicillin G with the acyl intermediate of the TEM-1 β-Lactamase. The broken bond between the carbonyl carbon and the nitrogen of the β-Lactam ring is shown with the grey dashed line. [PDB: 1FQG] (Nitrogen - blue; Sulfur - yellow; Oxygen - red; Carbon - grey) |
The catalytic serine residue is deacylated and recovered from the reaction (Figure 6). The Glu166 residue’s carboxylate group accepts a proton from a water molecule. The resulting OH- molecule attacks the carbonyl carbon of the former β-Lactam ring, and the serine can be released. The proton taken by the Glu166 is transferred to the Ser70 Oˠ, thereby regenerating the initial catalyst (Strynadka et al., 1992).

|
| Figure 6. Condensed mechanism of deacylation in the presence of water. Tetrahedral intermediates and proton-carrying residues have been excluded for simplicity. Arrows indicate electron pushing. Structures were created using ChemDraw based on information presented in (Strynadka et al., 1992). |
Ambler C
Ambler class C β-Lactamases, referred to as AmpC enzymes, most notably hydrolyze cephalosporins and penams. The name “AmpC” connects these β-Lactamases across many bacterial species. However, the individual enzymes of this class generally do not have unique names and are not phylogenetically related (Jia et al., 2017). AmpC in E. coli resembles the class A β-Lactamases (Figure 7), with domain 1 consisting of a 9-stranded β-sheet flanked by 3 ⍺-helices. Domain 2 consists of 11 ⍺-helices (Usher et al., 1998). The active site lies between these two domains, and the binding site in AmpC is more open relative to other SBLs, consistent with AmpC’s ability to accommodate the bulkier side chains of cephalosporins (Jacoby, 2009).
![Figure 7. AmpC β-Lactamase enzyme of E. coli. Ser64 and Lys67 are part of the essential SXXK motif critical for enzymatic function. Tyrosine-150 contributes to stabilizing the deacylation reaction. [PDB ID: 3BLS] (Nitrogen - blue; Sulfur - yellow; Oxygen - red; Carbon - grey)](https://cdn.rcsb.org/rcsb-pdb/content/61b762c769569d045d35a99c/ampc.png)
|
| Figure 7. AmpC β-Lactamase enzyme of E. coli. Ser64 and Lys67 are part of the essential SXXK motif critical for enzymatic function. Tyrosine-150 contributes to stabilizing the deacylation reaction. [PDB ID: 3BLS] (Nitrogen - blue; Sulfur - yellow; Oxygen - red; Carbon - grey) |
The mechanism of action of AmpC is similar to that of the other SBLs such as TEM-1 discussed above. AmpC has the conserved sequence motif SXXK as well. In E. coli, the motif is represented as: Ser64, Val65, Ser66, and Lys67; Ser64 is the catalytic residue used in the acylation step (Usher et al., 1998). The major difference in AmpC compared to other SBLs is that there is much debate about the mechanisms of deacylation. While the mechanism of deacylation is still being investigated, it is known that the residue Tyrosine-150 is involved (Figure 8).
Acylation occurs just as it does in TEM-1. Serine-64, which has been deprotonated by Lys67, attacks and forms a covalent bond with the carbonyl group of the β-Lactam ring; this results in cleavage of the amide bond of the β-Lactam ring and an acyl-enzyme intermediate is formed.
![Figure 8. Acyl-enzyme intermediate of AmpC from E. coli in complex with the cephalosporin Cefotaxime. The β-Lactam nitrogen serves as the proton acceptor in activating the catalytic water and also helps stabilize the deacylation intermediate along with Tyrosine-150. [PDB ID: 4KG2] (Nitrogen - blue; Sulfur - yellow; Oxygen - red; Carbon - grey)](https://cdn.rcsb.org/rcsb-pdb/content/61b762c769569d045d35a99c/ampc-mechoverview.png)
|
| Figure 8. Acyl-enzyme intermediate of AmpC from E. coli in complex with the cephalosporin Cefotaxime. The β-Lactam nitrogen serves as the proton acceptor in activating the catalytic water and also helps stabilize the deacylation intermediate along with Tyrosine-150. [PDB ID: 4KG2] (Nitrogen - blue; Sulfur - yellow; Oxygen - red; Carbon - grey) |
The deacylation mechanism involves using a catalytic water molecule to react with the acyl-enzyme intermediate just like with TEM-1. While the details of this mechanism are still being investigated, the result is the same: the enzyme is regenerated and the drug is broken. In AmpC, it is proposed that the nitrogen of the β-Lactam ring accepts a proton from water, similar to how glutamic acid-166 did in TEM-1. This activates the catalytic water molecule which attacks the acyl-enzyme intermediate. The nearby Tyrosine-150 residue and β-Lactam nitrogen stabilize the resulting deacylation intermediate (Chen et al., 2006).
Regulation of AmpC
AmpC-mediated drug resistance is also unique from other SBLs because of how it is expressed. While AmpC is the β-Lactamase that destroys the β-Lactam drug, there are 3 additional enzymes involved in the regulation of AmpC expression which ensure that high enough levels of the β-Lactamase are expressed at the right times. AmpG is an inner cell membrane permease protein. AmpD is an amidase enzyme that catalyzes the hydrolysis of amide bonds. AmpR is a transcription factor whose conformation can activate or suppress the expression of the Amp genes on bacterial RNA (Lister, Wolter, and Hanson, 2009).
In wild-type Pseudomonas aeruginosa strains, basal expression of the AmpC enzyme occurs, (Figure 9), which means it is expressed at a level that does not cause high levels of resistance to β-Lactam drugs. The peptidoglycan cell wall goes through a recycling process in which muropeptides are released from the cell wall layer. These units are transported into the cytoplasm through AmpG permease. These muropeptide molecules are cleaved by AmpD amidase, generating free tripeptides which will be used to make new cell wall precursors, specifically UDP-MurNAc-pentapeptides. UDP-MurNAc-pentapeptide interacts with AmpR and causes AmpR to retain its conformation, preventing efficient transcription of AmpC genes. This results in basal, or low levels, AmpC expression (Lister, Wolter, and Hanson, 2009).
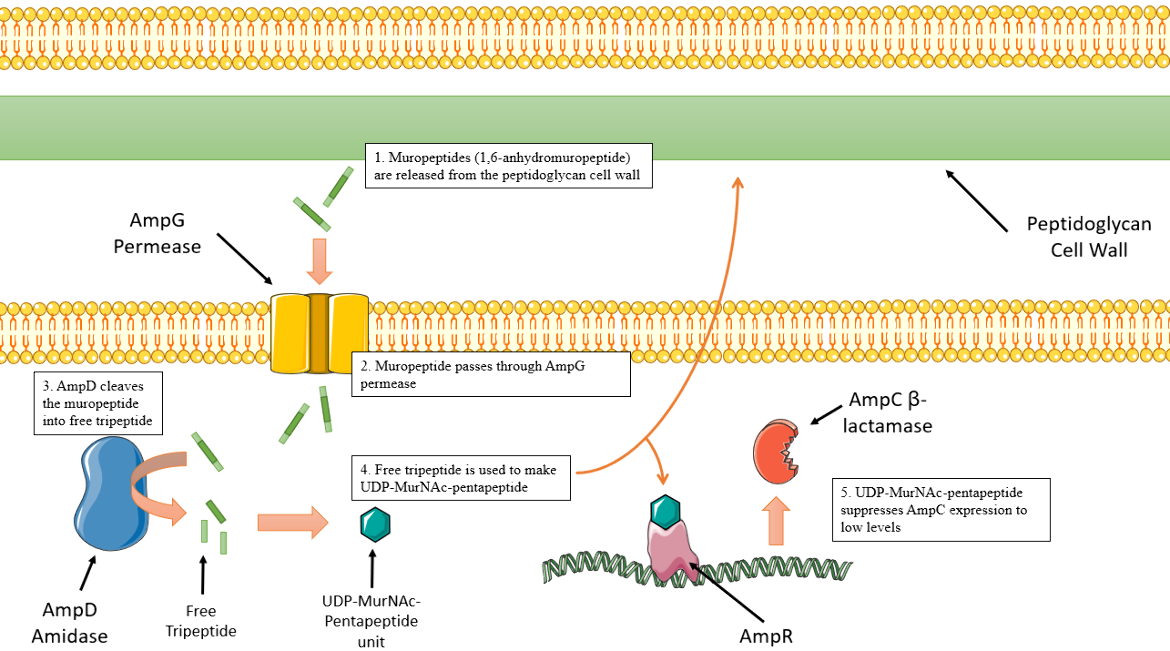
|
| Figure 9. Depiction of AmpC basal expression sequence. Cartoon created using Servier Medical Art. (Lister, Wolter, and Hanson, 2009). |
AmpC expression can be “induced” or stimulated in certain cellular conditions, specifically in the presence of β-Lactam drugs. β-Lactams like Cefoxitin or Imipenem cross through the outer cell membrane and inhibit late-stage peptidoglycan cell wall biosynthesis. This results in increased levels of muropeptide that AmpD cannot process fast enough. Thus, the muropeptides are able to interact more with AmpR than the UDP-MurNAc units. The muropeptides interact with AmpR and convert AmpR into a transcriptional activator, the opposite effect of the UDP-MurNAc units. As a result, more AmpC is expressed (Figure 10), and a higher level of resistance to the β-Lactams is observed. When there is less drug in the periplasmic space, the pools of muropeptide also decrease, and AmpD sufficiently processes these units and AmpC expression returns to basal levels once again (Lister, Wolter, and Hanson, 2009).
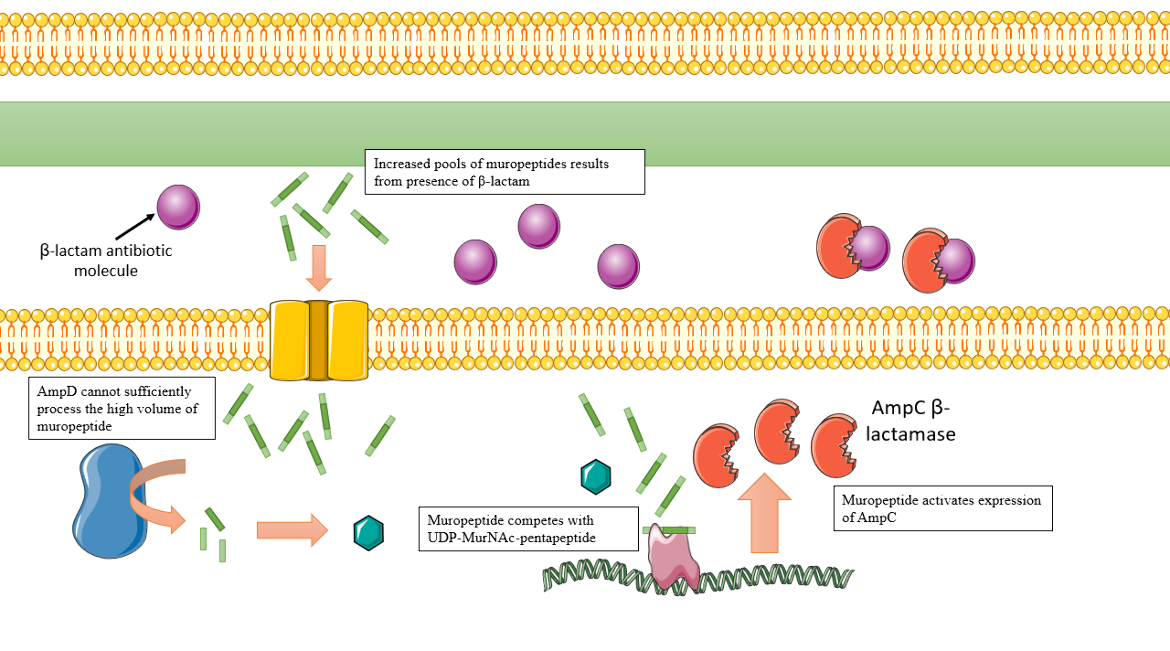
|
| Figure 10. Depiction of the AmpC overexpression process, which results in β-Lactam resistance. Cartoon created using Servier Medical Art. (Lister, Wolter, and Hanson, 2009). |
Mutations in AmpD, or decreased AmpD expression cause the pool of muropeptides to remain high, even in non-inducing conditions. As a result, AmpR is locked in its activating conformation and ampC is constantly expressed at high levels even when β-Lactams are not present. This process produces populations of bacteria with high levels of resistance to β-Lactams like Cefoxitin or Imipenem (Lister, Wolter, and Hanson, 2009).
Ambler D
Ambler class D β-Lactamases are the final class of serine β-Lactamases. They share less than 20% amino acid sequence identity with the class A and C SBLs. Additionally, many class D enzymes hydrolyze oxacillin and cloxacillin faster than classical penicillins, earning class D enzymes their additional name of oxacillinases (Sun et al., 2003). The mechanism of oxacillinases is similar to that of the other serine β-Lactamases.
To understand the critical difference that class D enzymes have mechanistically from the other SBLs, OXA-1, the most common class D β-Lactamase, will be used for the discussion.
Table 2. OXA-1 Data: Drug classes that OXA-1 β-Lactamase successfully resists and the bacterial species that possess genes expressing OXA-1. Information obtained from CARD.
| Drug Classes | Bacterial Species |
|---|---|
| Penam, Cephalosporin | Citrobacter freundii, Citrobacter koseri, Enterobacter aerogenes, Enterobacter asburiae, Enterobacter cloacae, Enterobacter hormaechei, Enterobacter kobei, Escherichia coli, Klebsiella oxytoca, Klebsiella pneumoniae, Morganella morganii, Proteus mirabilis, Providencia rettgeri, Pseudomonas aeruginosa, Raoultella planticola, Salmonella enterica, Serratia marcescens, Shigella dysenteriae, Shigella flexneri, Shigella sonnei |
In brief, the protein structure of OXA-1 consists of two domains (Figure 11) - domain one is a mixed domain consisting of a 6-stranded antiparallel β-sheet with helices on both sides, and domain two is all helical. The active site where the β-lactam binds resides in between these two domains. The active site contains the critical motif Ser67, Thr68, Phe69, and modified Lys70 (sequence from OXA-1 in E. coli) with Ser67 being the catalytic residue for acylation. OXA-1 uses similar stabilizing interactions and mechanisms as the enzymes in classes A and C. The primary difference is the use of a carboxylated lysine side chain in the active site as a reaction intermediate (Sun et al., 2003).
![Figure 11. The OXA-1 β-Lactamase with the modified (carboxylated) lysine residue and catalytic serine residue are shown in magnified view. [PDB: 1M6K, Sun et al., 2003] (Nitrogen - blue; Oxygen - red; Carbon - grey)](https://cdn.rcsb.org/rcsb-pdb/content/61b762c769569d045d35a99c/oxaoverview.png)
|
| Figure 11. The OXA-1 β-Lactamase with the modified (carboxylated) lysine residue and catalytic serine residue are shown in magnified view. [PDB: 1M6K, Sun et al., 2003] (Nitrogen - blue; Oxygen - red; Carbon - grey) |
At pH of 6 and above, the Lys70 residue is carboxylated, and the carboxylate group deprotonates the catalytic Ser67 which will go on to attack the β-Lactam carbonyl carbon in the same fashion as the other serine β-Lactamases in the acylation step (Figure 12 and 13).
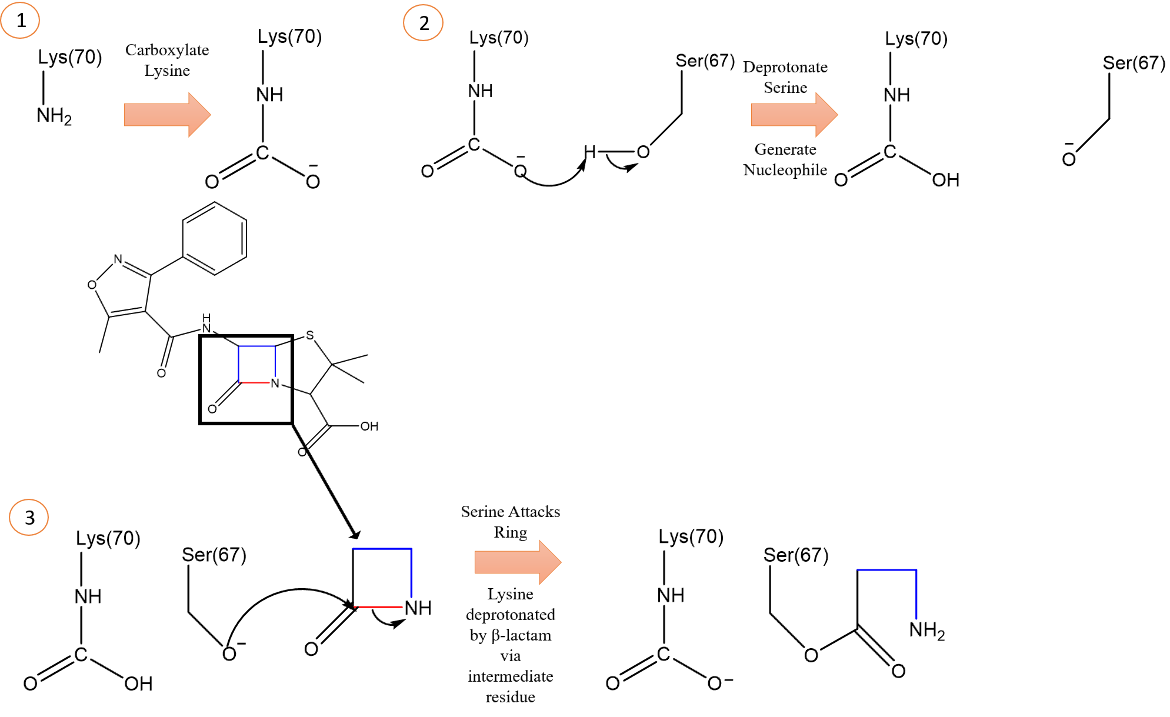
|
| Figure 12. Condensed mechanism of the acylation step forming the acyl-intermediate in which OXA-1 is still attached to the β-Lactam drug Oxacillin. Some tetrahedral intermediates and proton-carrying residues have been excluded for simplicity. Arrows indicate electron pushing. The structures were created using ChemDraw. |
![Figure 13. Structure of the acyl intermediate, with an isolated view of the now broken drug Oxacillin on the left. The broken amide bond is shown as a gray dashed line. [PDB: 4MLL] (Nitrogen - blue; Oxygen - red; Sulfur - yellow; Carbon - grey).](https://cdn.rcsb.org/rcsb-pdb/content/61b762c769569d045d35a99c/oxa1-wdrug.png)
|
| Figure 13. Structure of the acyl intermediate, with an isolated view of the now broken drug Oxacillin on the left. The broken amide bond is shown as a gray dashed line. [PDB: 4MLL] (Nitrogen - blue; Oxygen - red; Sulfur - yellow; Carbon - grey). |
Additionally, the carboxylate group of the carboxylated lysine takes a proton from a catalytic water molecule, and the resulting hydroxide ion will deacylate the drug molecule (Figure 14). The same proton accepted by the lysine carboxylate group is donated back to Ser67 to regenerate the catalyst. These different proton transfers in the acylation and deacylation steps are accommodated by the carboxylated Lys70. However, in class A and C SBLs, two different residues are required to carry out these respective functions in acylation and deacylation (Sun et al., 2003).
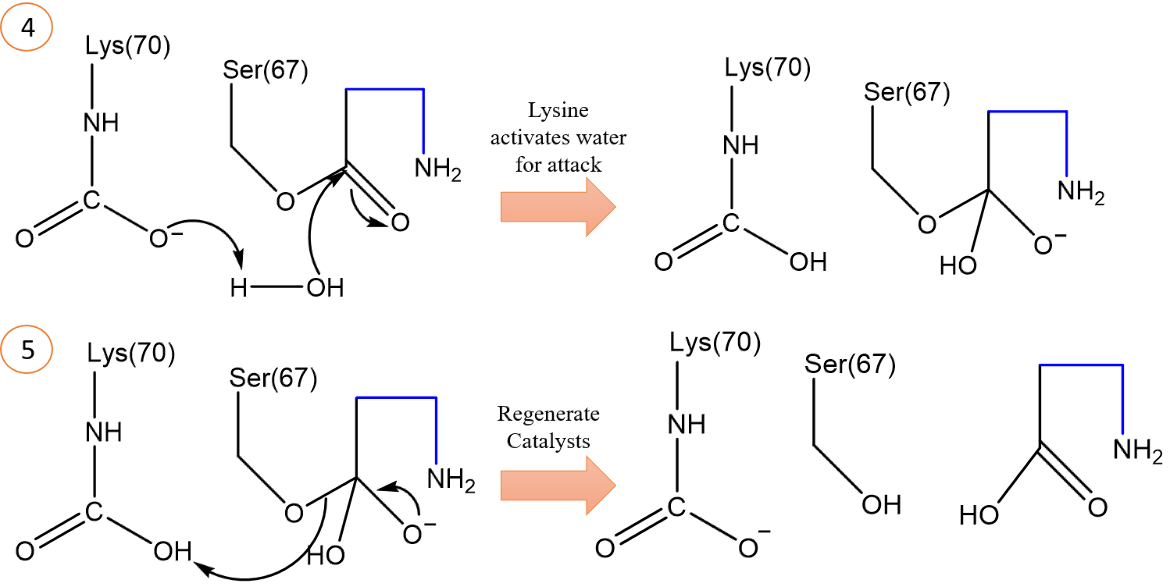
|
| Figure 14. Condensed mechanism of the deacylation step of Oxacillin hydrolysis. Some tetrahedral intermediates and proton-carrying residues have been excluded for simplicity. Arrows indicate electron pushing. The structures were created using ChemDraw. (Sun et al., 2003). |
Ambler B
Class B β-Lactamases, also referred to as metallo-β-Lactamases, use zinc ions to activate a water molecule and catalyze its addition to the β-Lactam ring to inactivate the drug (Figure 15), unlike their SBL counterparts which use their serine residue to attack and break the β-Lactam ring (Fernandes, Amador, and Prudêncio, 2013). Metallo-β-Lactamases have a fundamentally similar mechanism for the destruction of β-Lactams. To understand the differences, the New Delhi metallo-β-Lactamase-1 (NDM-1) will be the enzyme of reference. NDM-1 genes confer nearly complete resistance to almost all β-Lactam drugs, including carbapenems, in enteric bacteria. This is particularly concerning because carbapenems have a wide spectrum of activity and are often reserved for use against multi-drug resistant gram-negative bacteria. NDM-1 genes are typically found on transferable plasmids, prone to rearrangement and horizontal gene transfer. Thus, it comes as no surprise that after NDM-1’s discovery in India, it rapidly spread across the globe (King et al., 2012).
Table 3. NDM-1 Data: Drug classes that NDM-1 β-Lactamase successfully resists and the bacterial species that carry genes expressing NDM-1. Information obtained from CARD.
| Drug Classes | Bacterial Species |
|---|---|
| Penam, Cephamycin, Cephalosporin, Carbapenem | Acinetobacter baumannii, Acinetobacter nosocomialis, Citrobacter freundii, Enterobacter aerogenes, Enterobacter asburiae, Enterobacter cloacae, Enterobacter hormaechei, Enterobacter kobei, Escherichia coli, Klebsiella oxytoca, Klebsiella pneumoniae, Morganella morganii, Proteus mirabilis, Providencia rettgeri, Providencia stuartii, Pseudomonas aeruginosa, Raoultella planticola, Salmonella enterica, Serratia marcescens, Shigella sonnei, Vibrio parahaemolyticus |
In brief, NDM-1 has the ⍺β/β⍺ structure typical of most metallo-β-lactamases, with the zinc-containing active site near the edge of one of the β-sandwiches. NDM-1 has an active site cleft larger than most metallo-β-lactamases due to the orientations of two loops flanking the active site. This larger active site, along with its electrostatic profile, contributes to its increased range of β-lactam antibiotics that it can destroy.
![Figure 15. The protein structure of NDM-1, with a magnified view of the active site, is shown. Zn2+ ions and catalytic water are shown in relative position in the active site [PDB: 4EYF] (Nitrogen - blue; Oxygen - red; Sulfur - yellow; Carbon - grey).](https://cdn.rcsb.org/rcsb-pdb/content/61b762c769569d045d35a99c/ndm-1.png)
|
| Figure 15. The protein structure of NDM-1, with a magnified view of the active site, is shown. Zn2+ ions and catalytic water are shown in relative position in the active site [PDB: 4EYF] (Nitrogen - blue; Oxygen - red; Sulfur - yellow; Carbon - grey). |
Zinc ion 1 electrostatically orients the carbonyl oxygen of the drug to facilitate catalysis. Zinc ion 2 is involved in interactions with the amide nitrogen of the β-Lactam ring (Figure 16, 17). A hydroxide ion is generated by deprotonation of a catalytic water molecule by the basic histidine-120 residue. This hydroxide ion serves as a nucleophile and attacks the carbonyl carbon, similar to serine in the SBLs. This attack causes the amide bond to break and gives the nitrogen-leaving group on the former β-Lactam ring a negative charge. This negative charge is stabilized by zinc ion 2’s positive charge. The zinc ions are held closer to each other during the attack to ensure zinc ion 2’s proper location relative to the amide N. This is to ensure proper stabilization of the reaction intermediate. It is observed that the zinc-zinc distance increases following hydrolysis, which weakens the stabilization of the nitrogen-leaving group, allowing it to be protonated and leave the active site (King and Strynadka, 2011).
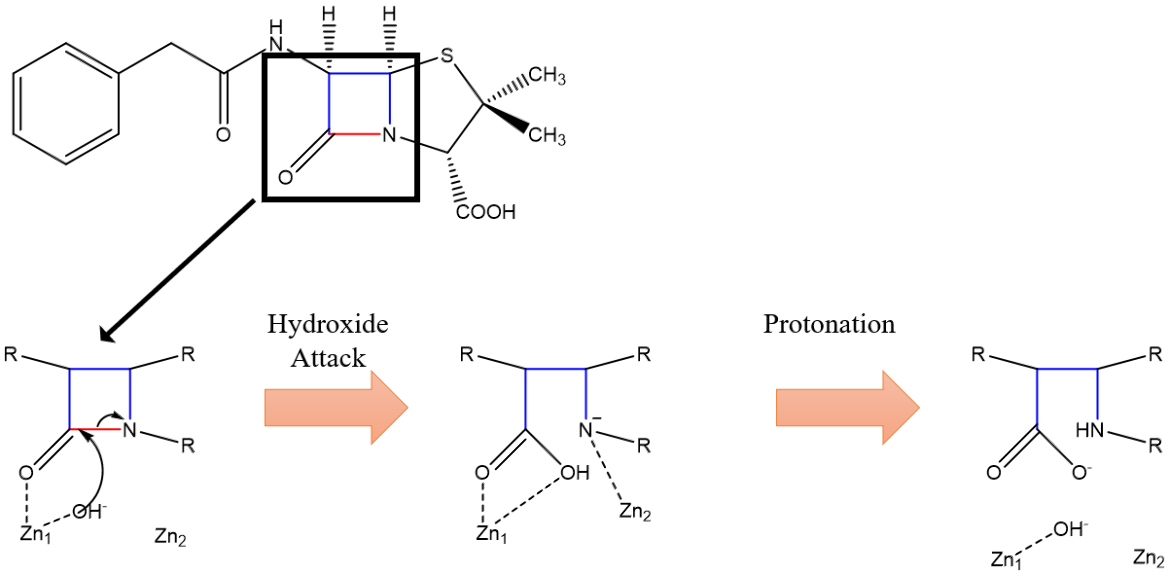
|
| Figure 16. Condensed mechanism of NDM-1 zinc-mediated hydrolysis of β-Lactam. Dashed lines indicate stabilizing interactions, not bonds. Arrows indicate electron pushing. (King and Strynadka, 2011). |
![Figure 17. The protein structure of NDM-1 is shown with a magnified view of the active site. Zn2+ ions, catalytic water, and hydrolyzed penicillin G are shown in relative position in the active site. The former amide bond (which is broken) is depicted with a dashed grey line [PDB: 4EYF] (Nitrogen - blue; Oxygen - red; Sulfur - yellow; Carbon - grey)](https://cdn.rcsb.org/rcsb-pdb/content/61b762c769569d045d35a99c/ndm-1-4eyf.png)
|
| Figure 17. The protein structure of NDM-1 is shown with a magnified view of the active site. Zn2+ ions, catalytic water, and hydrolyzed penicillin G are shown in relative position in the active site. The former amide bond (which is broken) is depicted with a dashed grey line [PDB: 4EYF] (Nitrogen - blue; Oxygen - red; Sulfur - yellow; Carbon - grey) |
Clinical Implications of Resistance by β-lactamases
Penicillin was the first antibiotic to be discovered and has found its place as one of the most used antibiotics in modern medicine. β-Lactams such as penicillin and carbapenems make up approximately 50% of all prescribed antibiotics (King, Worrall, Gruninger, & Strynadka, 2012). They stop cell wall synthesis in bacteria and provide a favorable means to treat infection because they have limited toxicity, exceptional bioavailability, and a relatively wide spectrum of activity. However, many β-Lactams have been rendered ineffective due to the evolution of functional β-Lactamases which inactivate these drugs. This has since become a major public health threat. Exploring the structures of β-Lactamases and understanding their mechanisms of action is critical for developing additional antibiotics that are effective in treatment but are not destroyed by β-Lactamases.
Currently, there are three main methods of combating β-Lactamase mediated resistance. First is the development of new β-Lactams that are not recognized by, and therefore safe from, the β-Lactamase enzymes and their destructive effects. The second is the use of entirely different types of antibiotics that are not β-Lactams. The third method is the coadministration of β-Lactamase inhibitors which inactive the enzymes before they get a chance to destroy the antibiotic, allowing for effective drug administration (King & Strynadka, 2011). The β-Lactamases of Ambler class A are generally inhibited by clavulanic acid, tazobactam, or sulbactam, and Ambler class C enzymes are poorly inhibited by these compounds (Munita & Arias, 2016). Ambler C enzymes are inhibited by cloxacillin, oxacillin, and aztreonam (Jacoby, 2009). Ambler class D SBLs are poorly inhibited by clavulanic acid (Munita & Arias, 2016), however, they are inhibited by NaCl (Sun et al., 2003). Due to the unique mechanism of Ambler class B enzymes, inhibitors like clavulanic acid or tazobactam do not work, however, ion-chelating agents such as ethylenediaminetetraacetic acid (EDTA) can be used for inhibition (Munita & Arias, 2016). These methods can only finitely prolong the utility of β-Lactams.
References
Ambler, R. (1980) The Structure of β-Lactamases. Philosophical Transactions of the Royal Society B: Biological Sciences 289, 321-331. https://doi.org/10.1098/rstb.1980.0049
Bush, K., Bradford, P. (2016) β-Lactams and β-Lactamase Inhibitors: An Overview. Cold Spring Harbor Perspectives in Medicine 6, a025247. https://doi.org/10.1101/cshperspect.a025247
Chen, Y., Minasov, G., Roth, T., Prati, F., Shoichet, B. (2006) The Deacylation Mechanism of AmpC β-Lactamase at Ultrahigh Resolution. Journal of the American Chemical Society 128, 2970-2976. https://doi.org/10.1021/ja056806m
Fernandes, R., Amador, P., Prudêncio, C. (2013) β-Lactams. Reviews in Medical Microbiology 24, 7-17. https://doi.org/10.1097/MRM.0b013e3283587727
Fonzé, E., Charlier, P., To'th, Y., Vermeire, M., Raquet, X., Dubus, A., Frère, J. (1995) TEM1 β-lactamase structure solved by molecular replacement and refined structure of the S235A mutant. Acta Crystallographica Section D Biological Crystallography 51, 682-694. https://doi.org/10.1107/s0907444994014496
Hall, B., Barlow, M. (2005) Revised Ambler classification of β-lactamases. Journal of Antimicrobial Chemotherapy 55, 1050-1051. https://doi.org/10.1093/jac/dki130
Jacoby, G. (2009) AmpC β-Lactamases. Clinical Microbiology Reviews 22, 161-182. https://doi.org/10.1128/cmr.00036-08
Jia et al. (2017). CARD 2017: expansion and model-centric curation of the Comprehensive Antibiotic Resistance Database. Nucleic Acids Research 45, D566-573. https://doi.org/10.1093/nar/gkw1004
Kelly, J. A., Dideberg, O., Charlier, P., Wery, J. P., Libert, M., Moews, P. C., Knox, J. R., Duez, C., Fraipont, C., Joris, B. (1986). On the origin of bacterial resistance to penicillin: comparison of a beta-lactamase and a penicillin target. Science (New York, N.Y.), 231(4744), 1429–1431. https://doi.org/10.1126/science.3082007
King, D., King, A., Lal, S., Wright, G., Strynadka, N. (2015) Molecular Mechanism of Avibactam-Mediated β-Lactamase Inhibition. ACS Infectious Diseases 1, 175-184. https://doi.org/10.1021/acsinfecdis.5b00007
King, D., Strynadka, N. (2011) Crystal structure of New Delhi metallo-β-lactamase reveals molecular basis for antibiotic resistance. Protein Science 20, 1484-1491. https://doi.org/10.1002/pro.697
King, D., Worrall, L., Gruninger, R., Strynadka, N. (2012) New Delhi Metallo-β-Lactamase: Structural Insights into β-Lactam Recognition and Inhibition. Journal of the American Chemical Society 134, 11362-11365. https://doi.org/10.1021/ja303579d
Lister, P., Wolter, D., Hanson, N. (2009) Antibacterial-Resistant Pseudomonas aeruginosa: Clinical Impact and Complex Regulation of Chromosomally Encoded Resistance Mechanisms. Clinical Microbiology Reviews 22, 582-610. https://doi.org/10.1128/cmr.00040-09
Munita, J., Arias, C. (2016) Mechanisms of Antibiotic Resistance. Virulence Mechanisms of Bacterial Pathogens, Fifth Edition 481-511. https://doi.org/10.1128/microbiolspec.vmbf-0016-2015
Paterson, D., Bonomo, R. (2005) Extended-Spectrum β-Lactamases: a Clinical Update. Clinical Microbiology Reviews 18, 657-686. https://doi.org/10.1128/cmr.18.4.657-686.2005
Strynadka, N., Adachi, H., Jensen, S., Johns, K., Sielecki, A., Betzel, C., Sutoh, K., James, M. (1992) Molecular structure of the acyl-enzyme intermediate in β-lactam hydrolysis at 1.7 Å resolution. Nature 359, 700-705. https://doi.org/10.1038/359700a0
Sun, T., Nukaga, M., Mayama, K., Braswell, E., Knox, J. (2003) Comparison of β-lactamases of classes A and D: 1.5-A crystallographic structure of the class D OXA-1 oxacillinase. Protein Science 12, 82-91. https://doi.org/10.1110/ps.0224303
Tipper, D. (1985) Mode of action of β-lactam antibiotics. Pharmacology & Therapeutics 27, 1-35. https://doi.org/10.1016/0163-7258(85)90062-2
Tipper, D. J., Strominger, J. L. (1965). Mechanism of action of penicillins: a proposal based on their structural similarity to acyl-D-alanyl-D-alanine. Proceedings of the National Academy of Sciences of the United States of America, 54(4), 1133–1141. https://doi.org/10.1073/pnas.54.4.1133
Usher, K., Blaszczak, L., Weston, G., Shoichet, B., Remington, S. (1998) Three-Dimensional Structure of AmpC β-Lactamase from Escherichia coli Bound to a Transition-State Analogue: Possible Implications for the Oxyanion Hypothesis and for Inhibitor Design†. Biochemistry 37, 16082-16092. https://doi.org/10.1021/bi981210f
Ruiz J. Etymologia: TEM. Emerg Infect Dis. 2018 Apr [date cited]. https://doi.org/10.3201/eid2404.ET2404
April 2025, Sameer Ahmad, Helen Gao; Reviewed by Dr. James R. Knox
https://doi.org/10.2210/rcsb_pdb/GH/AMR/drugs/amr-mech/blmase
